戦略提携室
株式会社理研イノベーションとの連携のもと、当プログラムが推進する創薬テーマ・プロジェクトについて、企業や大学、外部研究機関との共同研究やライセンシング等のアライアンス部分を担当しています。
先端計算科学基盤ユニットでは分子動力学専用計算機MDGRAPE-4Aにより得られるタンパク構造情報を生かしたインシリコ創薬方法論を開発・構築・研究応用しています。
先端計算科学基盤ユニットでは、分子動力学計算に特化した専用計算機MDGRAPE-4A(1マイクロ秒/day(10万原子系)、スパコンの5倍相当)を2019年に完成させました(図1)。現在、この分子動力学専用スパコンを使用し、タンパク質の機能解明や創薬への応用の研究を行なっています。
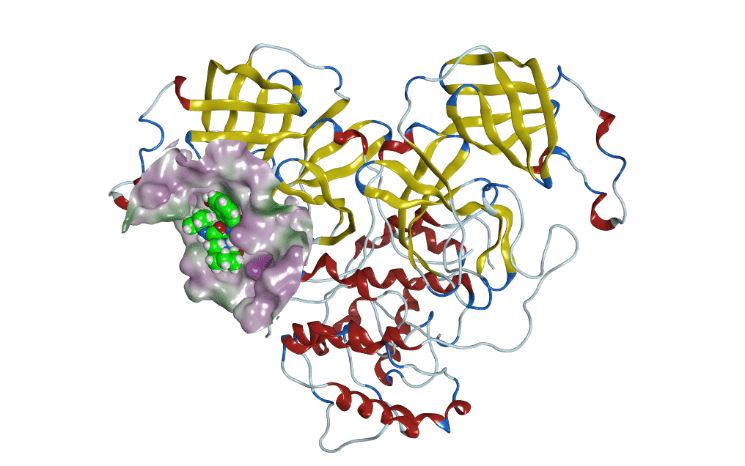
その成果の一つとして、世界に先駆けてSARS-CoV-2のメインプロテアーゼの分子動力学シミュレーションデータを公開し、更に、その薬物結合に伴うポケット構造変化の観測に成功しました(Komatsu, T. S. et al. 2020図2) 。
他のスパコンシステム(外部リンク)と連携しながら、創薬研究を推進・加速化するインシリコ創薬方法論を開発・構築し研究応用しています。

クリックするとYouTubeへ移動します
インシリコ創薬は医薬品開発序盤の薬物探索(大規模な化合物データベースの中から創薬標的分子に対し薬理活性を持つ化合物を選出)と薬物最適化(選出された化合物を化学修飾し薬理活性・ADMETを更に改善)に適用されます。
インシリコ創薬の技術はバーチャルな薬物構造式を用いて実施できるという大きな利点を持ち、近年の計算技術の進歩による精度や速度の飛躍的向上に伴って創薬現場において必須な技術として期待されています。
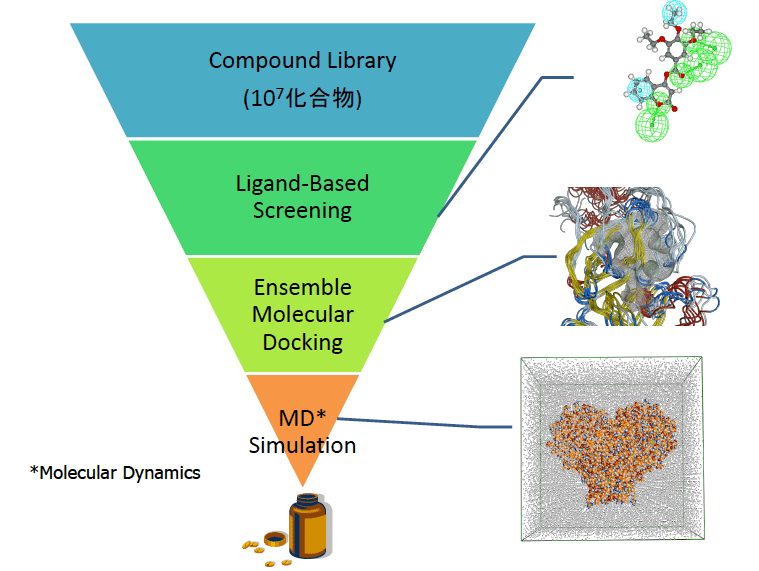
先端計算科学基盤では上記の分子動力学専用計算機(MDGRAPE-4A)活用した創薬手法の1つとして、分子ドッキングと分子動力学シミュレーションを組み合わせたインシリコスクリーニング手法を開発し、創薬研究に適用してきています。薬物探索の工程では、複数の探索方法を階層的に組み合わせて、膨大な化合物候補を創薬標的タンパク質に適した薬物群へと絞り込んでいきます(図3参照)。
探索序盤は、精度はあまり高くないものの、高速に大規模な薬物ライブラリから創薬標的に適した薬物群への絞り込みを実行できるという利点を持つLigand Based Drug Design (LBDD)を実施しています。次に、LBDDよりも計算コストは高いものの、その精度の信頼度は高い分子ドッキング法(創薬標的タンパク質の作用部位(ポケット)と薬物の立体構造を考慮し、その複合体構造とその結合親和性を予測します)を実施しています。この際、先端計算科学基盤ユニットでは、実験構造に加えて分子動力学シミュレーションによって得られる複数のタンパク質構造を使用したアンサンブルドッキングを実施します。
最終的には、膨大な分子動力学シミュレーションを使用したスクリーニングで候補薬物を絞っていきます。分子動力学シミュレーションでは、分子の動きと溶媒効果を考慮した高精度な結合親和性予測を実現できますが、その膨大な計算コストの為に薬物スクリーニングへ適用することは非常に困難とされてきました。スパコンなどの高性能計算機使用することで現実的な計算時間で高精度な分子動力学シミュレーションによるスクリーニングを実現しています。
近年、低分子医薬品向けのタンパク質は調査し尽くされ、これらの開発は閉塞的な状況にあると言われています。この問題点を解決するため、先端計算科学基盤ユニットでは、標的タンパク質上の隠れた薬物作用点を発見し、薬物設計を加速する新たな分子シミュレーション技術を整備しています。
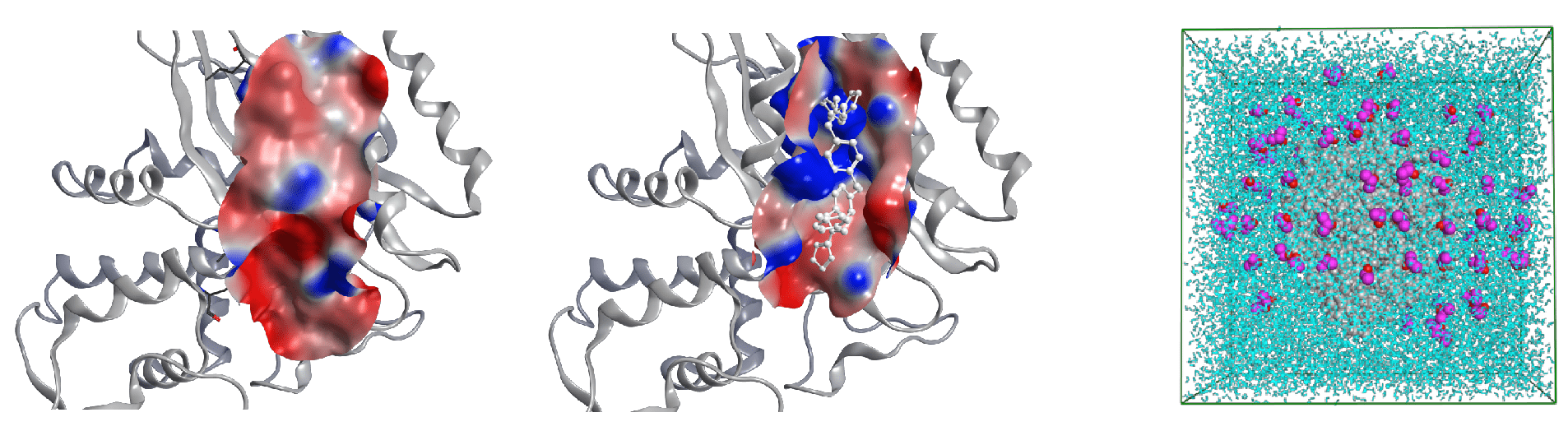
タンパク質―タンパク質間相互作用部位などは、構造柔軟性が高く、アポタンパク質の実験構造から潜在的な薬物結合ポケットを発見できない場合があることが知られています(図4)。そこで、共溶媒(低分子有機化合物と水分子の混合溶媒)中のタンパク質の分子動力学シミュレーションによって、アポタンパク質表面の潜在的な薬物結合ポケットを見つけ出します。これによって、創薬タンパク質の新しい薬物作用点での低分子薬物開発を実施することができます。
インシリコスクリーニングにより選出された候補化合物は実験的に評価されて、ヒット化合物となります。これらのヒット化合物は、更なる化学修飾を施され、より強い薬理活性化合物へと洗練されていきます。ヒット化合物の化学修飾においては、インシリコスクリーニング以上にタンパク質―化合物間の高精度な結合親和性予測がもとめられるため、先端計算科学では、そのための計算手法の研究開発を試みています。
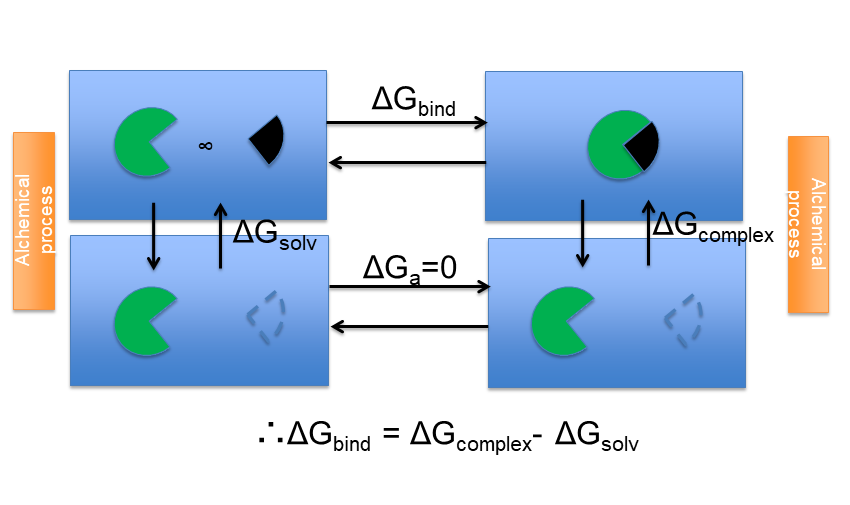
代表的な計算法として、非物理的経路の熱力学サイクルを用いて高精度に結合親和性を予測できるアルケミカル自由エネルギー計算法の流れを以下に示す(図5)。今後、様々な標的タンパクで評価検討例を蓄積しながら計算手法のさらなる改良を図る予定です。