戦略提携室
株式会社理研イノベーションとの連携のもと、当プログラムが推進する創薬テーマ・プロジェクトについて、企業や大学、外部研究機関との共同研究やライセンシング等のアライアンス部分を担当しています。
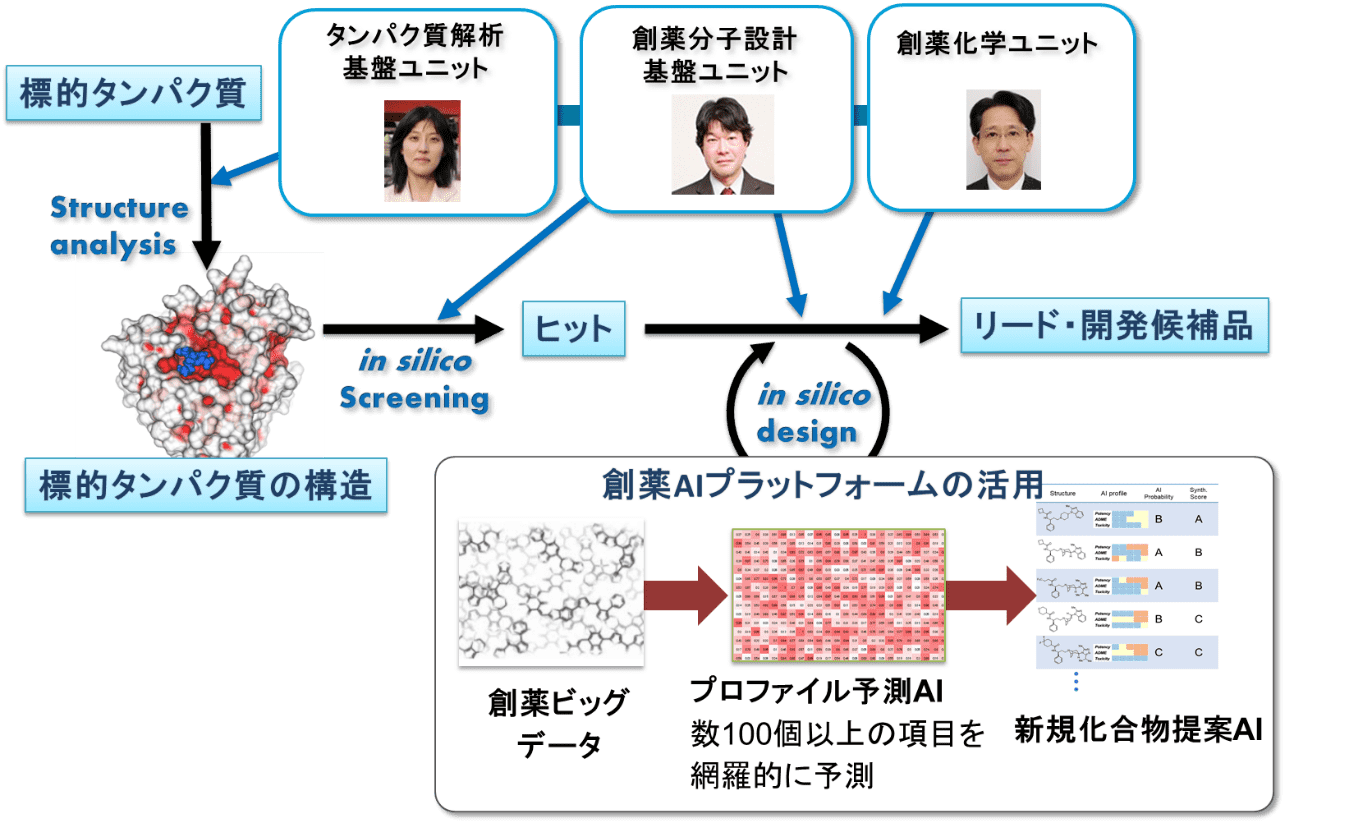
新しい創薬シーズの探索には、タンパク質の立体構造に基づく薬物設計(SBDD)が有用ですが、従来のドッキングでは、タンパク質の構造の自由度を考慮したり、ドッキング条件の検討を充分に行うことなく、本番のインシリコスクリーニングを行うために、特に難しい標的の場合には失敗に終わる場合もあります。創薬分子設計基盤ユニットでは、標的タンパク質の立体構造と既知阻害剤情報を最大限活かした高精度なインシリコスクリーニングのシステム(図1)を構築しています。
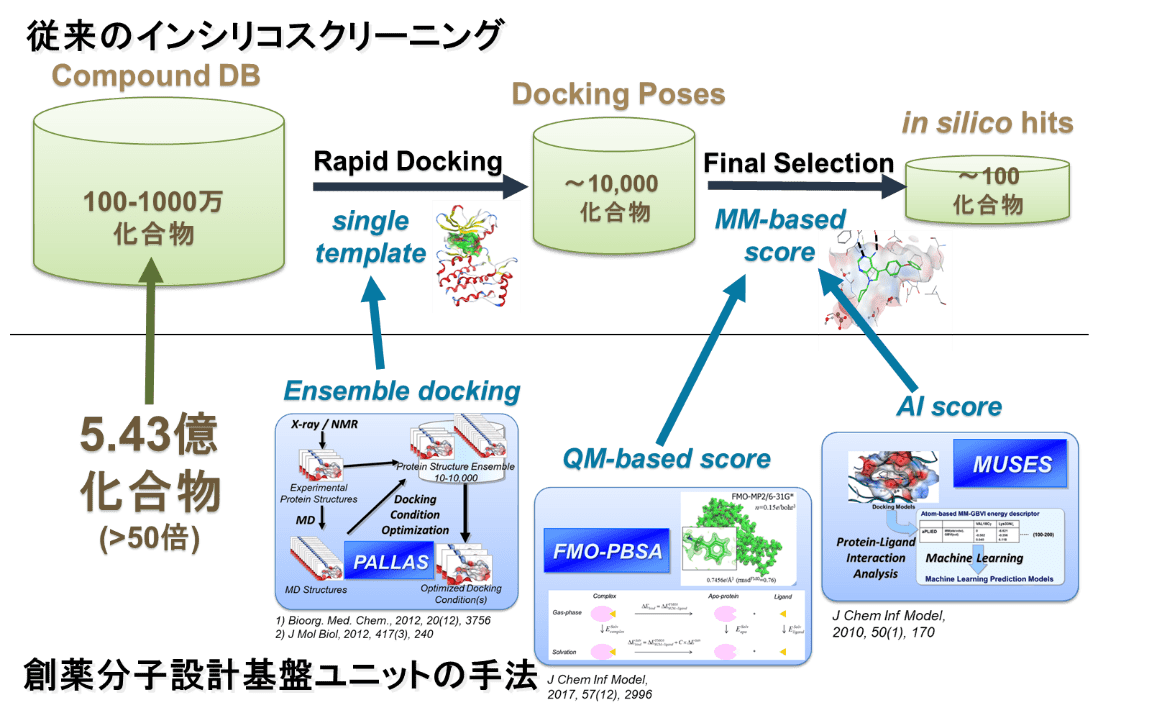
PALLASは、半自動的なドッキング条件の最適化システムであり、タンパク質の構造バリエーションや溶媒分子の有無など、様々な条件をインプットすると、多様なリガンドのドッキングに適したタンパク質構造セットを選び出します。選び出した最適条件を用いてドッキングすることによってドッキングの効率を向上させ、失敗を回避することができます。
PALLASによって選択した化合物から、最終的にアッセイするさらに少数の化合物を絞り込むために、タンパク質-リガンド間相互作用を原子単位で記述する新規パラメータ(aPLIED)を用いて活性化合物と不活性化合物(検証時は囮で代用します)を判別するAI予測システム(MUSES)を用います。このMUSESシステムは、既知の活性化合物と囮化合物をドッキングさせ、活性化合物と囮化合物の相互作用パターンのわずかな違いを統計解析によって検出することを可能とします。創薬分子設計基盤ユニットにおけるaPLIEDと呼ばれる新しい相互作用記述子(パラメータ)とSupport Vector Machineによる機械学習予測モデルは、市販のドッキングスコア(GLIDE score)や文献公知の記述子(PLIF)使った場合に比べて精度が大きく向上します。
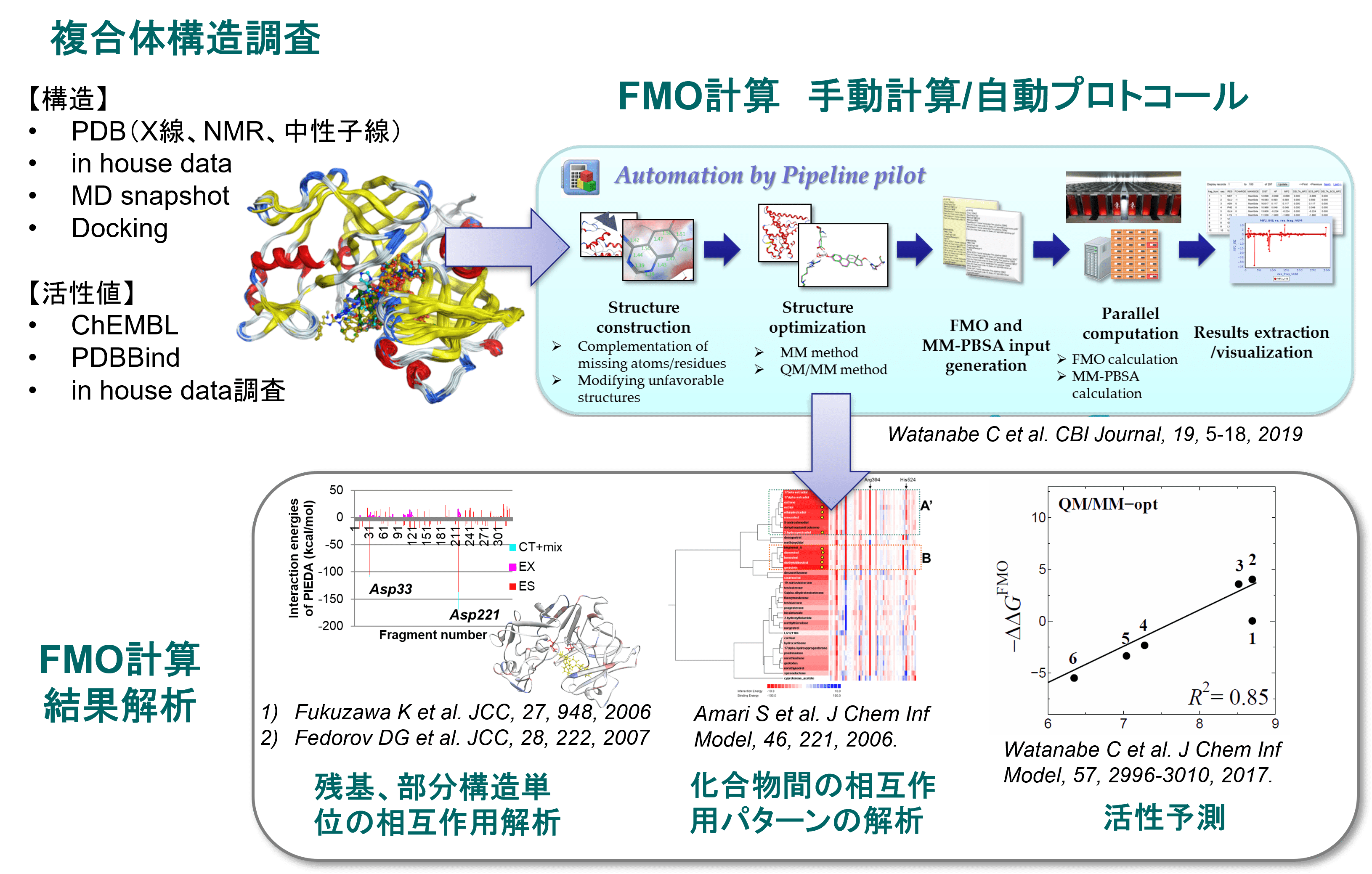
また、分子シミュレーションに基づく高精度の活性予測としては、タンパク質のような高分子に対する第一原理量子化学計算を可能にするFMO法と溶媒効果予測を組み合わせたFMO-PBSA法とFMOとAIを組み合わせた予測手法を開発しています。量子化学計算(FMO法)に基づく創薬基盤技術を図2に示します。
次の画像をクリックすると拡大します
創薬研究においては、創薬ターゲットへの活性だけではなく、オフターゲット、薬物動態(ADME)、毒性(T)のプロファイルを向上させることが必須であり、多くの場合創薬ターゲットに対する活性を獲得するよりも何倍も困難な作業であるために、創薬研究初期の律速段階となっています。創薬分子設計基盤ユニットでは、AMEDの次世代創薬AI事業(DAIIA)と連携して、膨大な創薬データと最新のAI技術に基づくAIプラットフォームを利用して、創薬分子設計を展開します。