Drug Discovery Molecular Simulation Platform UnitDrug Design Utilizing High-Performance Computers
This unit develops, constructs, and applies in-silico drug design strategies utilizing protein structure information obtained by MDGRAPE-4A, a special-purpose computer system for molecular dynamics simulations.
Ⅰ The special-purpose computer system for molecular dynamics simulations, MDGRAPE-4A
In 2019, this unit completed the MDGRAPE-4A (see Figure 1), a special-purpose computer system for molecular dynamics simulations (the achieved performance is roughly 1 microsecond per day for the systems including about 100 thousand atoms.).
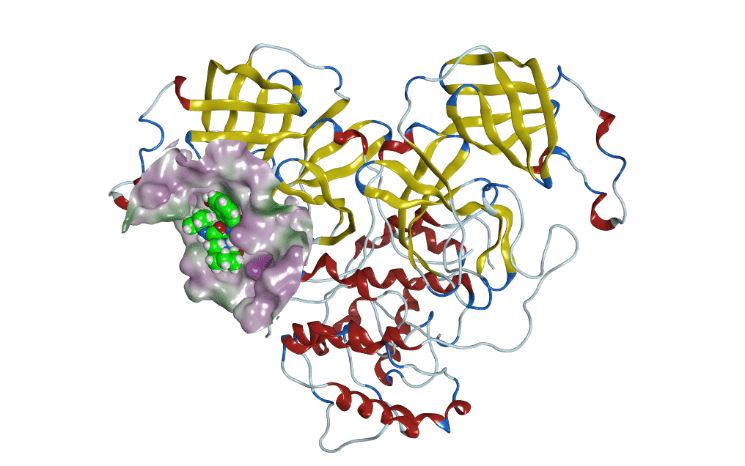
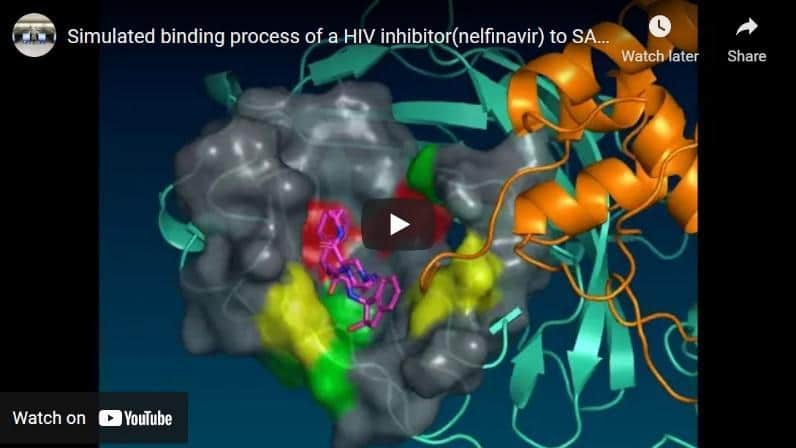
Currently, we are using this supercomputer dedicated to molecular dynamics to conduct research on protein functions and applying it to drug discovery. As one of our results, we were the first in the world to release the molecular dynamics simulation data of the main protease of SARS-CoV-2, and we succeeded in observing the behavior of the pocket conformational changes associated with its drug binding (Komatsu, T. S. et al., 2020; Figure 2). In collaboration with other supercomputers, the Unit is developing, constructing, and applying in-silico drug design strategies that promote and accelerate drug discovery research.
Figure 1. MDGRAPE-4A system and MDGRAPE-4A boardFigure 2. The structure of the HIV inhibitor (nelfinavir) bound SARS-CoV-2 main protease obtained from molecular dynamics simulation using a special-purpose computer system for molecular dynamics simulations, MDGRAPE-4A system.
II. Computational drug discovery using high-performance computers
Computational drug discovery approaches are applied to the compound screening process and the compound optimization. The computational drug discovery has the great advantage of being able to conduct research on virtual drug structures, and it has become an essential technology in drug development with the progress of computational technology in recent years.
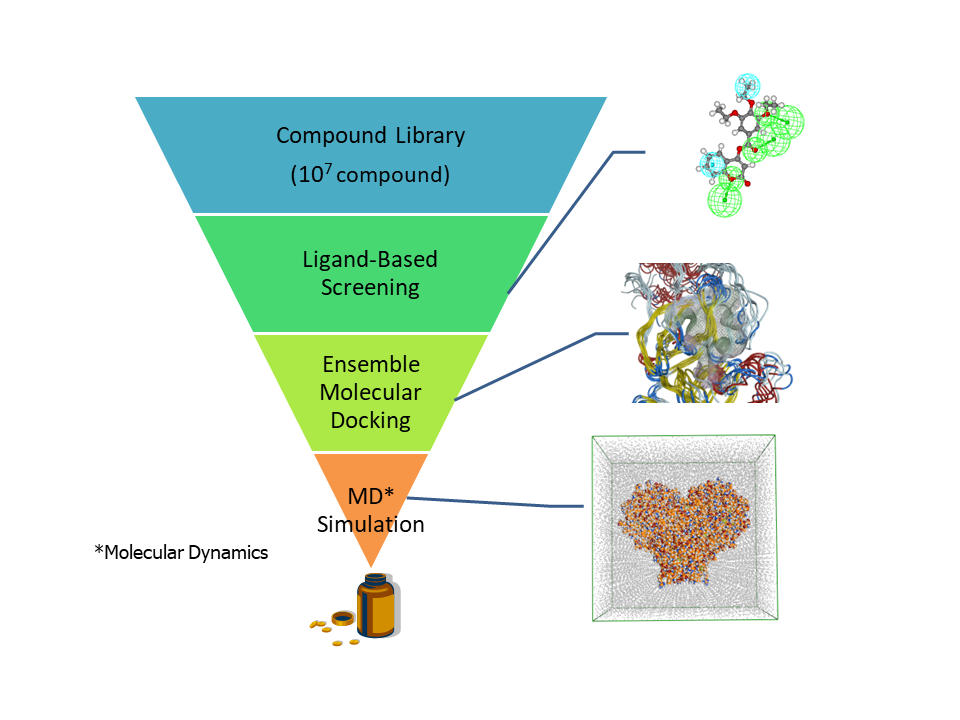
We have developed the compound screening method that combine molecular docking and molecular dynamics simulations utilizing high-performance computers (MDGRAPE-4A and so on). In the early stage of drug development, novel promising compounds are narrowed down from large compound libraries by using various computational methods (Figure 3).
In the initial phase of compound screening, ligand-based drug design (LBDD) is applied. LBDD has the advantage of fast filtering from large compound libraries to suitable compounds for a target protein, but it is not so accurate because they do not consider the structural information of the target protein. In the next phase, molecular docking that consider not only ligand structure but also protein structure is applied. Molecular docking is a more reliable method although it is more computationally expensive than LBDD. At this time, our unit performs ensemble docking that utilizes the various protein conformations obtained from molecular simulations as well as experimental conformations.
In the final stage, we apply molecular dynamics simulations to protein-ligand complex structures obtained from molecular docking and select compound candidates. These molecular dynamics simulations can consider the protein dynamics in solution and predict reliable ligand binding affinities, but the computational cost is very enormous. By using high performance computers, we realize compound screening by using molecular dynamics simulation in a realistic calculation time.
Figure 3. A flow of compound screening using molecular dynamics simulations in this unit.
III. Discovery of novel druggable site using high performance computers
In recent years, it is considered that target proteins for small molecule drugs have been depleted. To solve this problem, this unit has developed new molecular simulation techniques to discover hidden druggable pocket on target proteins and accelerate drug discovery.
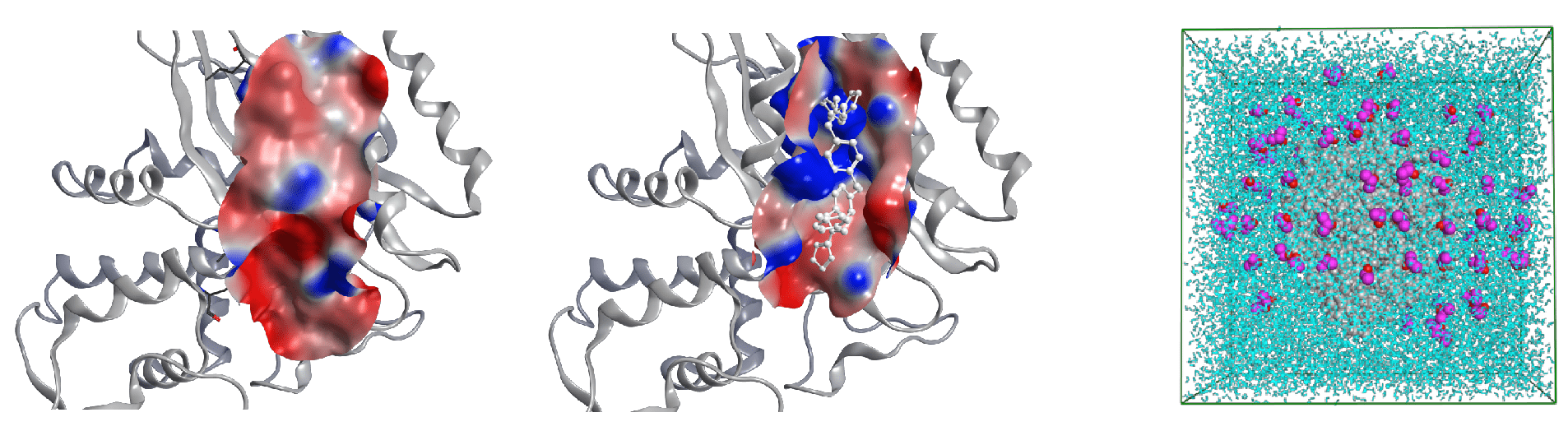
The target proteins involve the regions on protein−protein interactions or allosteric effect; since these regions lack apparent binding pockets, these were, for a long time, considered undraggable regions due to high structural flexibility (Figure 4). This unit has performed molecular dynamics simulation of apo protein using Mixed-solvent (for example, water-benzene mixture) to discover the hidden druggable pockets. These simulations expand the potential for small molecule drug development at new draggable sites.
Figure 4. Experimental structure example of hidden drug pocket, TEM-1 β-lactamase: In ligand unbound protein structure (left figure), the pocket conformation is not found, while the binding pocket is clearly formed in ligand bound protein structure (middle figure). The system of mixed-solvent molecular dynamics simulation system is shown in right figure. The water molecules, benzene molecules, and target protein are depicted in blue, purple and grey.
IV. Accurate protein-ligand binding affinity predictions (in this unit)
By experimental and computational compound screening, several hit compounds that bind to the target protein are found. These hit compounds are further improved to chemical compound with stronger pharmacological activity (compound optimization process). In this compound optimization process, more accurate binding affinity prediction of protein-ligand system is required, as compared with that of the compound screening mentioned above. This unit is developing the several computational methods for affinity prediction.
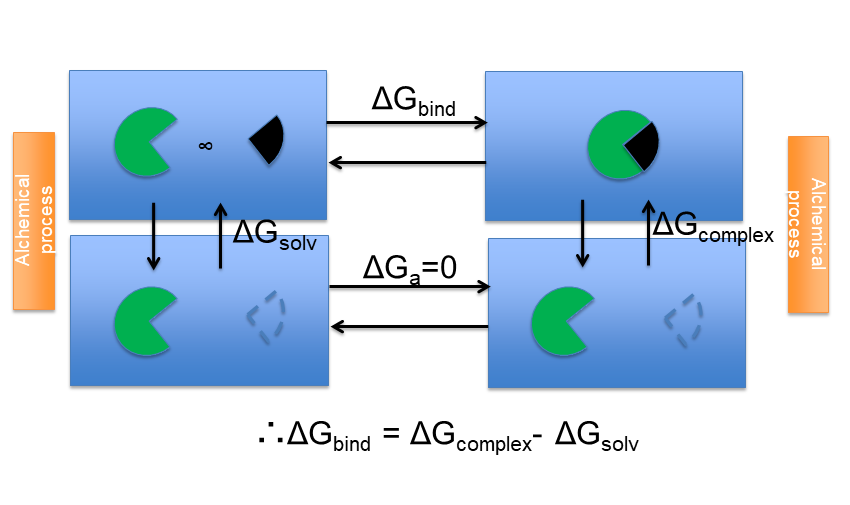
Among them, we prepared alchemical absolute free energy calculation method using the double decoupling scheme, which can give the accurate thermodynamic quantities (Figure 5). In the future, we plan to further improve the free energy calculation methods in drug discovery projects.
Figure 5. The thermodynamic cycle used in alchemical absolute free energy calculation. The target protein and ligand are depicted in green and black. The blue box contains the aqueous solution. The dashed line in the figure shows the state in which the ligand molecule is completely decoupled from the surrounding environment. The absolute binding free energy (ΔGbind) is computed from two free energy values (ΔGcomplex and ΔGsolv ).