Drug Discovery Computational Chemistry Platform UnitHigh-precision In Silico Screening Using the Latest Modeling Theories and Statistical Theories
High-precision in silico screening technologies
Drug design based on the three-dimensional structure of proteins (SBDD) is useful for searching
for new drug seeds. however, in conventional protein-ligand docking, the flexibility of protein
structure and other docking conditions were not sufficiently considered. These docking
conditions should be properly optimized for successful in silico screening. The Drug Discovery
Computational Chemistry Platform Unit has been constructing a highly accurate in silico
screening system (Figure 1.) that makes maximum use of the three-dimensional structure of the
target protein and information on known inhibitors.
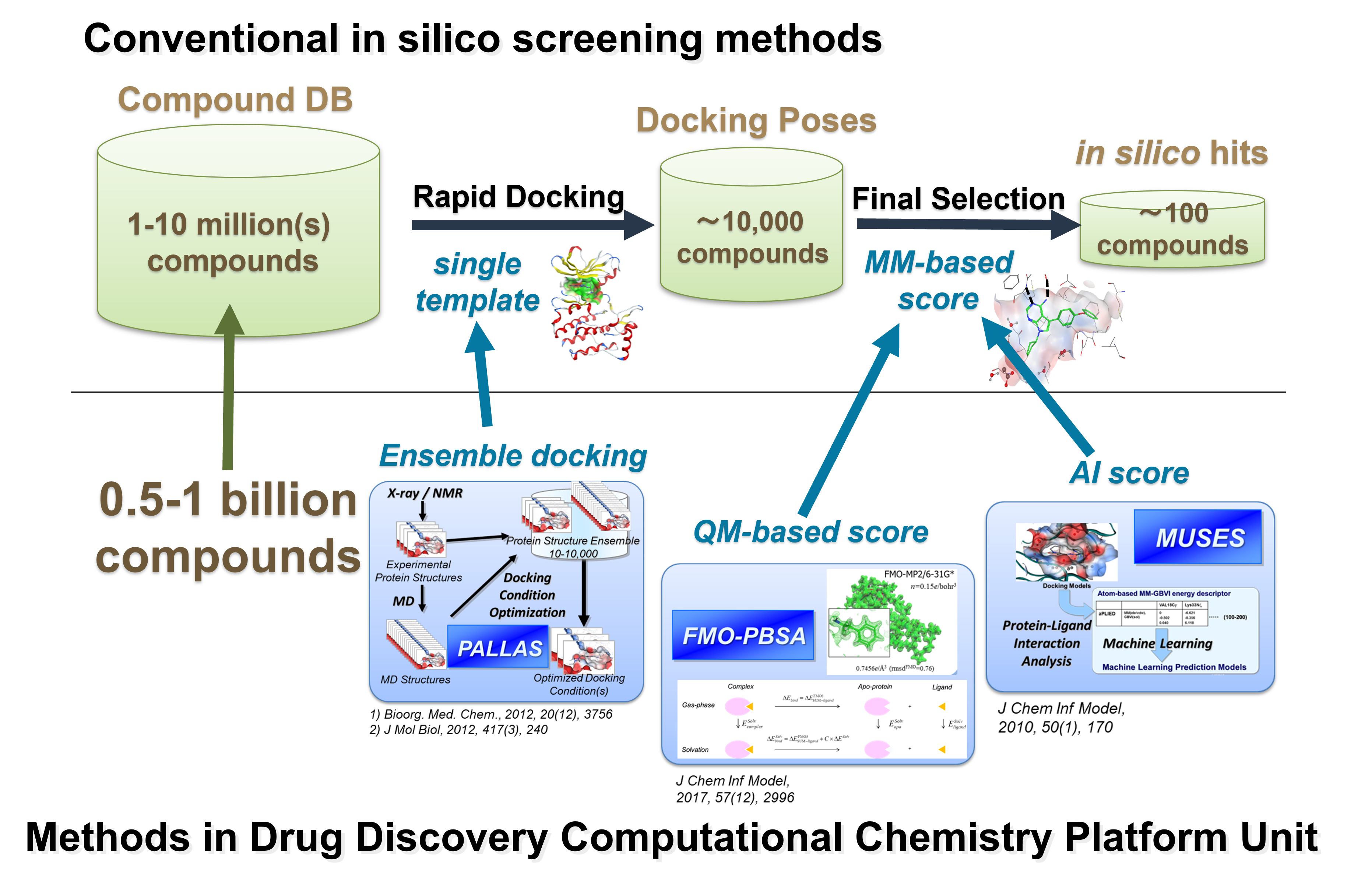
Figure 1. in silico screening system used in Drug Discovery Computational Chemistry
Platform Unit
PALLAS is a semi-automated docking condition optimization system that selects protein structure
sets and other docking settings suitable for detecting diverse ligands by examining many
conditions such as protein structural variations and the presence or absence of solvent
molecules. By docking using the selected optimum conditions, prediction performance can be
significantly improved.
For the final selection of compounds for biological assay, we employ Artificial Intelligence
(AI)-based prediction named MUSES using novel protein-ligand interaction descriptor named
aPLIED. The aPLIED provides protein-ligand interaction energy values atom-by-atom. The MUSES
system performs machine learning to detect subtle differences in the interaction pattern between
active and negative (decoy) compounds. The machine learning prediction model using aPLIED showed
significantly higher performance compared with a commercially available docking score (GLIDE
score) or a literature-known descriptor (PLIF).
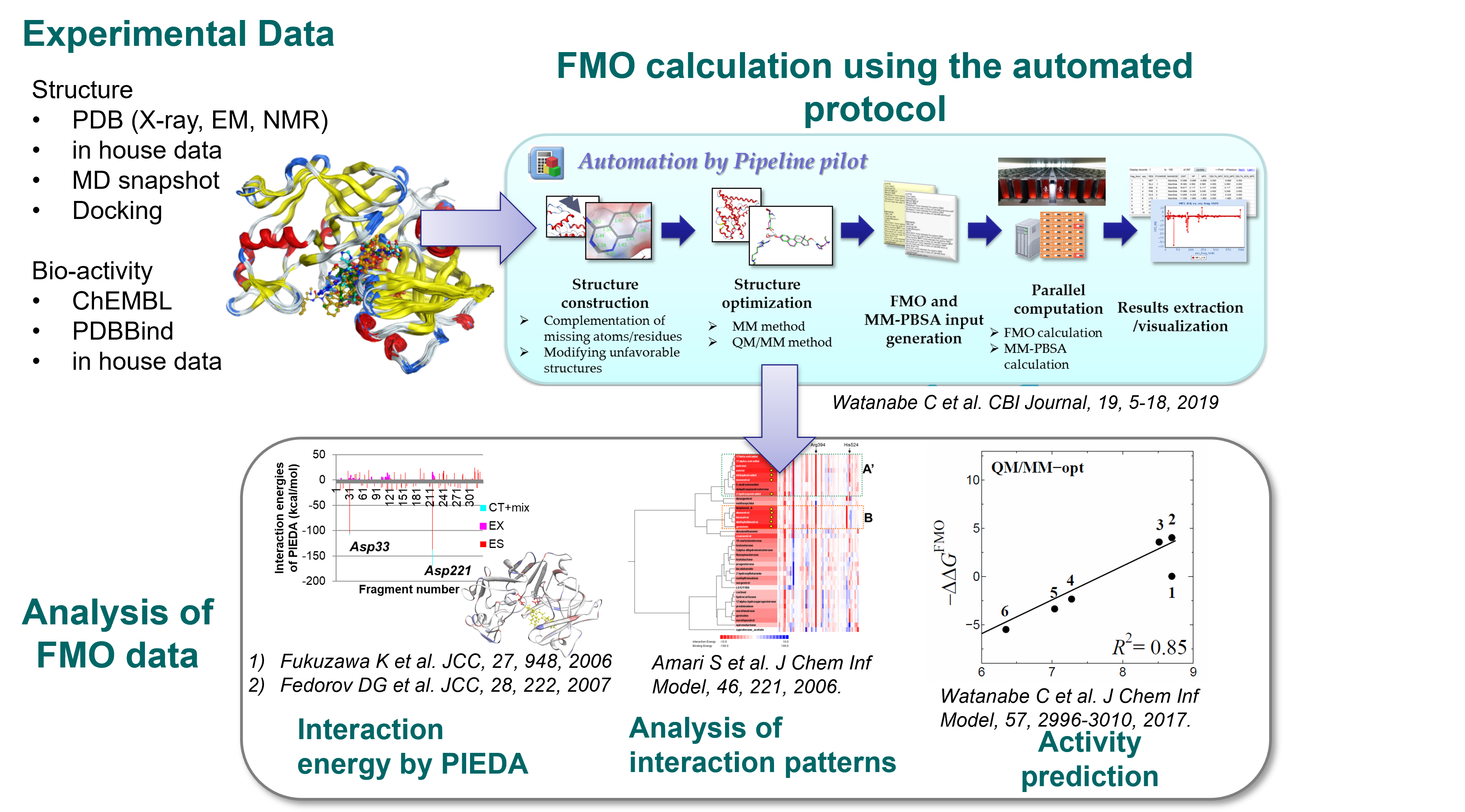
For highly accurate activity prediction based on molecular simulation, the FMO-PBSA method, which
combines the FMO method and solvent effect prediction. The FMO method enables first-principles
quantum chemistry calculations for proteins. We also combined FMO-based quantum chemical
interaction energy values with AI. Figure 2 shows the drug discovery technologies based on
quantum chemical calculation (FMO method).
Click on the following image to enlarge
Figure 2. Drug discovery technologies based on quantum chemical calculation (FMO
method)
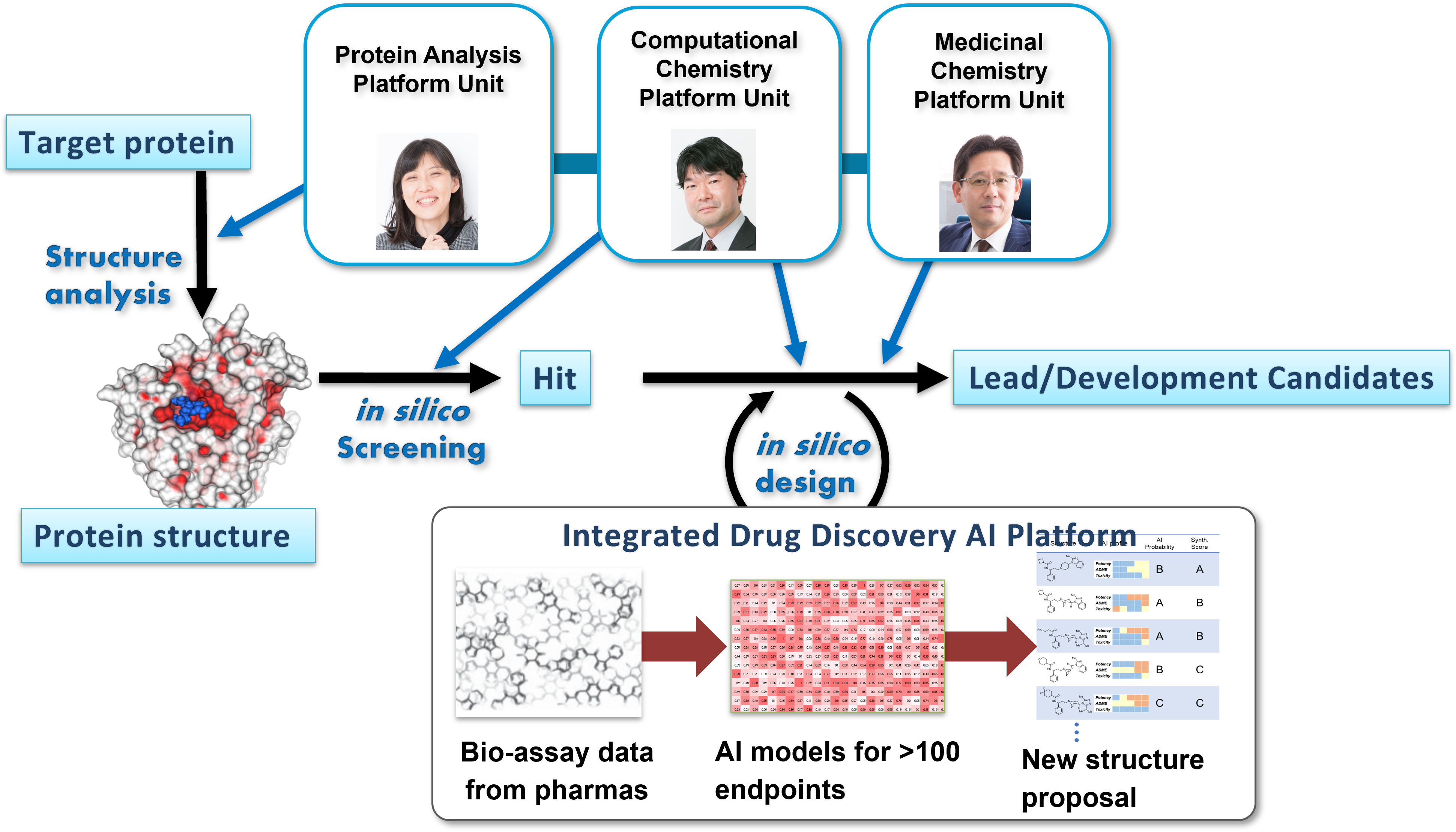
Molecular design from hit to lead/lead optimization
In drug discovery research, it is essential to improve not only the bioactivity for a target
protein but also off-targets, pharmacokinetic (ADME), and toxicity (T) profiles. Improvement of
the ADMET and off-target profile is a rate-determining stage in the early stages of drug
discovery. In collaboration with AMED's next-generation drug discovery AI project (DAIIA), our
platform unit develops integrated drug discovery AI platform including AI models for the
above-mentioned ADMET and off-target profile based on the latest AI technologies and learning
data provided from pharmaceutical companies and applied the AI models to real drug discovery
targets.
Figure 3. Drug design scheme collaborated among RIKEN DMP platform units