AI-driven Drug Discovery Collaborative UnitDevelopment of AI drug discovery technologies
Development of AI drug discovery technology by integrating " big data of drug
discovery with artificial intelligence"
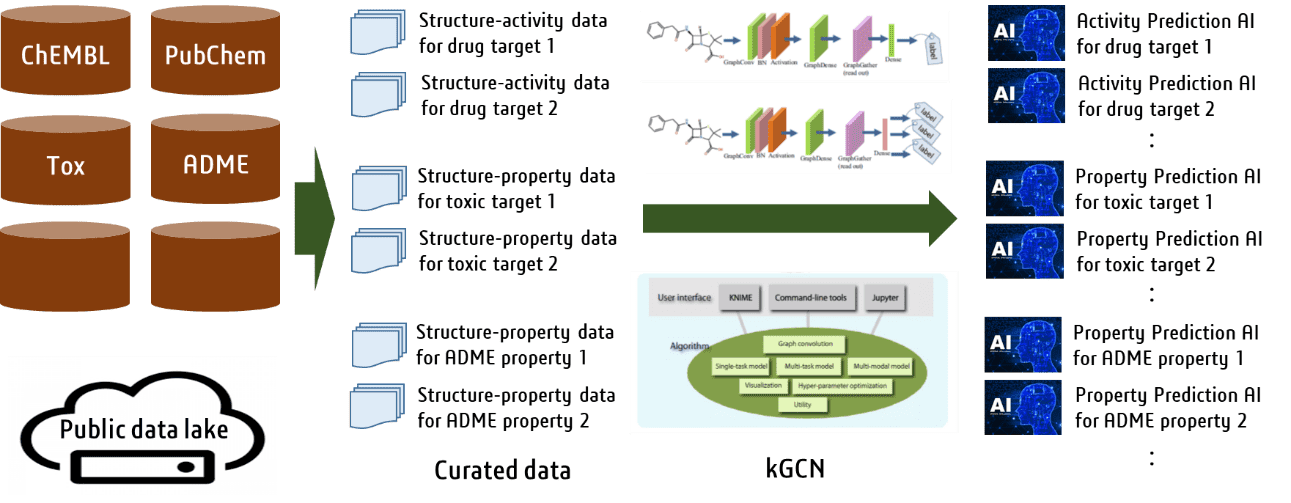
Numerous drug discovery databases, such as ChEMBL and PubChem, have been developed and made
publicly available, which provide data on activities of various drug discovery targets,
toxicity, and pharmacokinetics. With the graph convolutional neural networks, kGCN (J.
Cheminform., 2020, 12, 32), as the core developed at the Kyoto University, we are analyzing
these enormous amount of data and building various AI prediction systems.
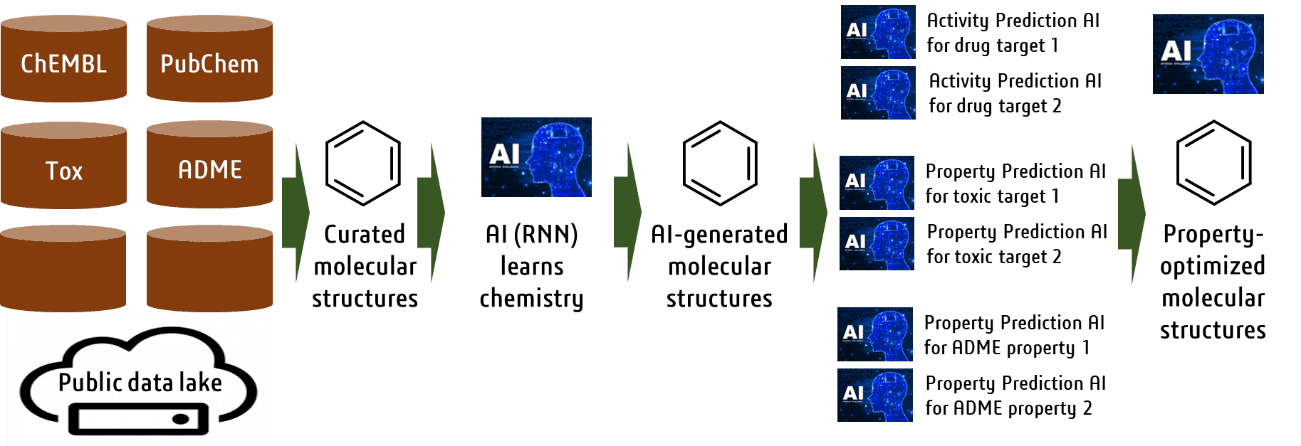
Small-molecule drugs have been designed by humans in most cases. To support drug design, we are
conducting research to make AI generate more diverse molecules with more desirable properties.
For this purpose, we have used an enormous number of chemical structures stored in public
databases to teach AI knowledge of chemistry for generating chemical structures. In addition, we
are developing AI to efficiently generate molecular structures with more desirable properties
using various prediction systems (J. Chem. Inf. Model., 2022, accepted).
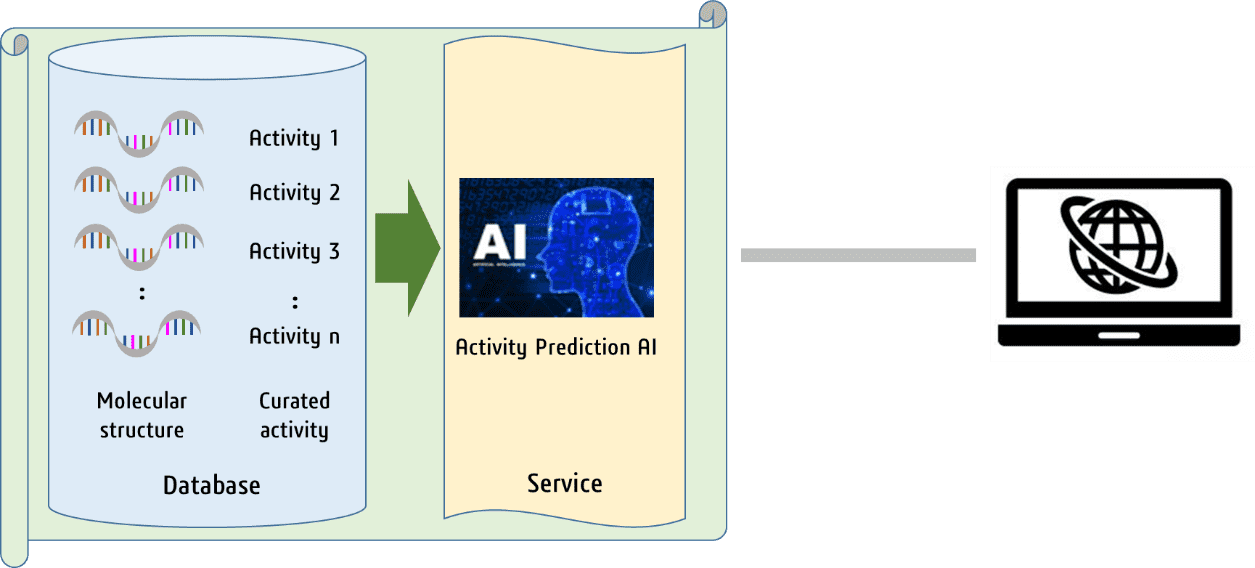
Nucleic acid drugs are attracting attention as a new modality, and several nucleic acid drugs
have been approved and used as drugs. We have created a database of nucleic acid sequences with
their activity and publish them for everyone’s free use (Nucleic Acid Res., 2021, 49, W193).
This system also incorporates an AI system that predicts the activity of any given nucleic acid
sequence.
Development of drug discovery technology through AI by combining simulations
with artificial intelligence
Drugs exert their pharmacological activity by binding to the target protein. Upon binding, water
molecules around the protein (hydrated waters) must be replaced by the drug. Therefore, the ease
of replacement of hydrated waters is very important for the drug to bind and exhibit its
activity.By using 3D-RISM theory, it becomes possible to estimate the hydration state of
proteins, which is critical for drug binding.
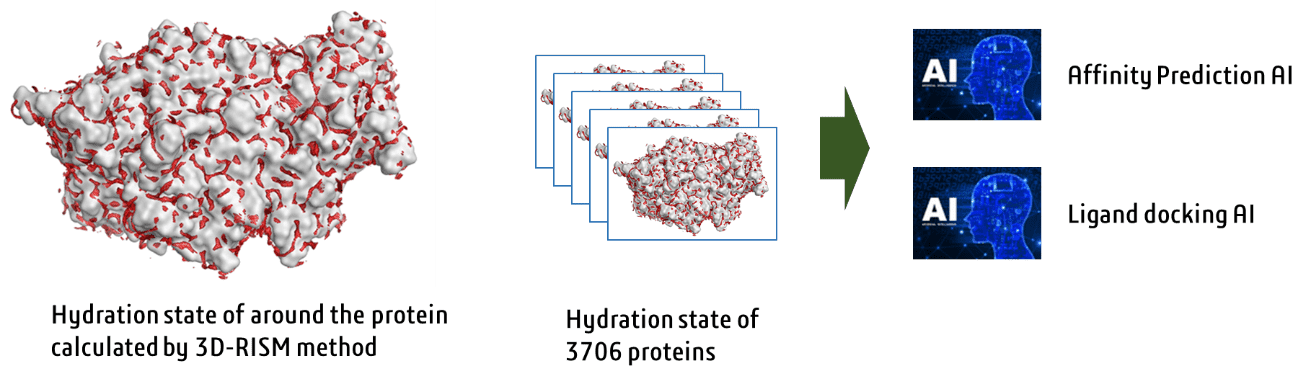
We used the supercomputers of RIKEN to comprehensively simulate the hydration state of 3,706
proteins using the 3D-RISM theory (J. Comput. Chem., 2020, 41, 2406). Based on the data of the
hydration states of 3,706 proteins, we have developed a methodology for quickly predicting the
hydration state of any proteins using AI (J. Chem. Inf. Model., 2022, 62, 4460). As a result,
3D-RISM calculations that used to take an average of several hours on a supercomputer can now be
completed in about tens of seconds by AI. As a further development, we are developing an AI that
uses hydration state information to predict binding affinities and correct binding modes.
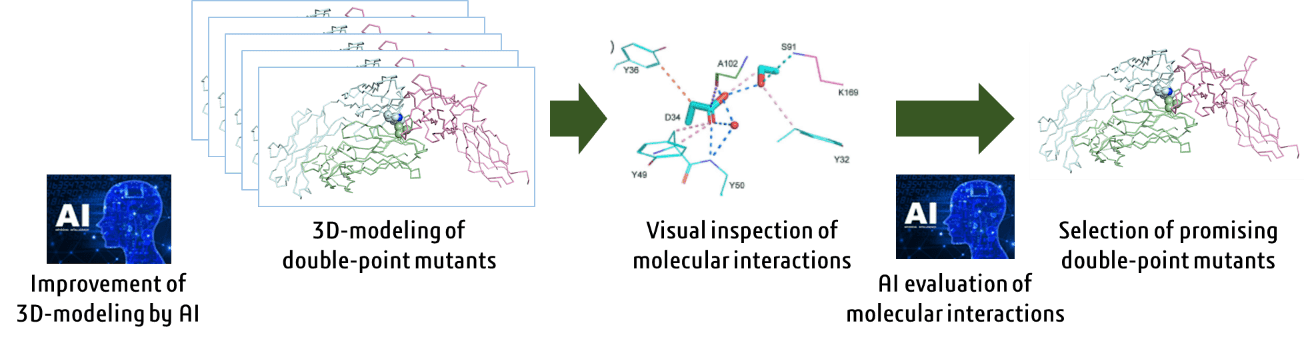
Antibody drugs are a source of innovative medicines. In developing antibody drugs, after
obtaining an active lead antibody, amino acids are mutated to improve its molecular properties
such as activity and stability. Normally, one amino acid is mutated; the changes in its activity
and properties are observed; and the good mutations are combined.
We have developed a new method of mutating two amino acids simultaneously (double-point mutant)
using simulation technologies (Scientific Reports, 2020, 10, 17590). In this method, a huge
number of 3D models of the double-point mutant should be generated and evaluated for improvement
of properties based on their 3D structures. Currently, generating 3D models of large number of
double-point mutants is automated; however, we are further refining and improving the method
using AI. The process of selecting mutants expected to improve activity from among numerous 3D
models has been done by researchers visually inspecting the 3D models, but we are working on
automating it with AI.