Virtual screening
ACPC
A pose prediction approach based on ligand 3D shape similarity
Molecular docking predicts the best pose of a ligand in the target protein binding site by sampling and scoring numerous conformations and orientations of the ligand. Failures in pose prediction are often due to either insufficient sampling or scoring function errors. To improve the accuracy of pose prediction by tackling the sampling problem, we have developed a method of pose prediction using shape similarity. It first places a ligand conformation of the highest 3D shape similarity with known crystal structure ligands into protein binding site and then refines the pose by repacking the side-chains and performing energy minimization with a Monte Carlo algorithm.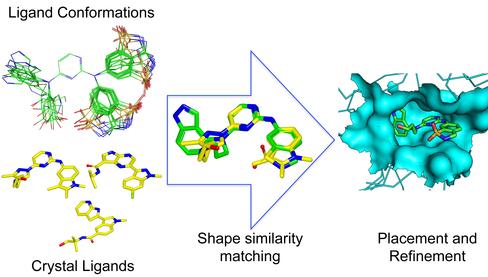
We have assessed our method utilizing CSARdock 2012 and 2014 benchmark exercise datasets consisting of co-crystal structures from eight proteins. Our results revealed that ligand 3D shape similarity could substitute conformational and orientational sampling if at least one suitable co-crystal structure is available. Our method identified poses within 2 Å RMSD as the top-ranking pose for 85.7 % of the test cases. The median RMSD for our pose prediction method was found to be 0.81 Å and was better than methods performing extensive conformational and orientational sampling within target protein binding sites. Furthermore, our method was better than similar methods utilizing ligand 3D shape similarity for pose prediction.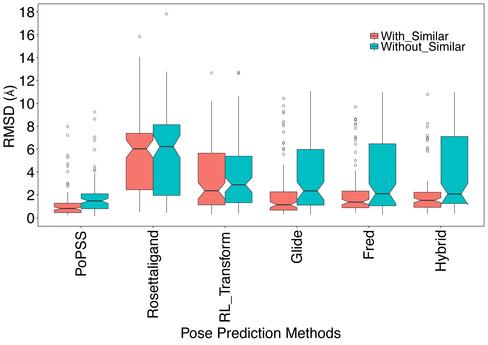
This work has been published in JCAMD (publication).
Molecular docking predicts the best pose of a ligand in the target protein binding site by sampling and scoring numerous conformations and orientations of the ligand. Failures in pose prediction are often due to either insufficient sampling or scoring function errors. To improve the accuracy of pose prediction by tackling the sampling problem, we have developed a method of pose prediction using shape similarity. It first places a ligand conformation of the highest 3D shape similarity with known crystal structure ligands into protein binding site and then refines the pose by repacking the side-chains and performing energy minimization with a Monte Carlo algorithm.
We have assessed our method utilizing CSARdock 2012 and 2014 benchmark exercise datasets consisting of co-crystal structures from eight proteins. Our results revealed that ligand 3D shape similarity could substitute conformational and orientational sampling if at least one suitable co-crystal structure is available. Our method identified poses within 2 Å RMSD as the top-ranking pose for 85.7 % of the test cases. The median RMSD for our pose prediction method was found to be 0.81 Å and was better than methods performing extensive conformational and orientational sampling within target protein binding sites. Furthermore, our method was better than similar methods utilizing ligand 3D shape similarity for pose prediction.
This work has been published in JCAMD (publication).