
Research
Variational Monte Carlo study for strongly correlated quantum systems
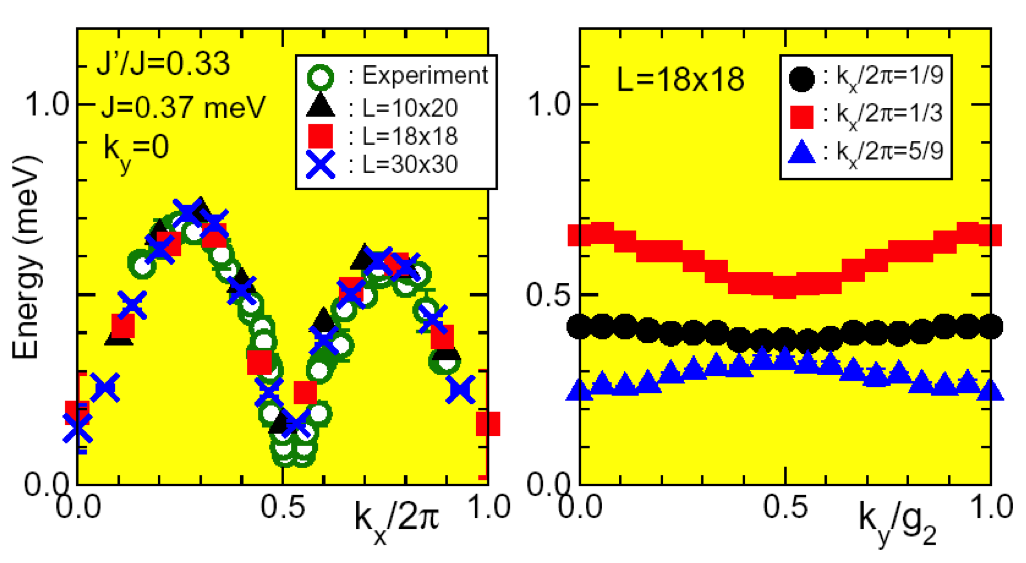 Variational Monte Carlo (VMC) method is a powerful tool to study the ground state properties of various strongly correlated quantum systems. In the VMC method, the appropriate trial wave function is assumed and its physical quantities are precisely calculated by Monte Carlo simulations within the statistical error. Because of the absence of the negative sign problem, the VMC method can be applied to many different classes of quantum systems including weakly as well as strongly correlated electrons, frustrated quantum magnets, and interacting bosons.
Variational Monte Carlo (VMC) method is a powerful tool to study the ground state properties of various strongly correlated quantum systems. In the VMC method, the appropriate trial wave function is assumed and its physical quantities are precisely calculated by Monte Carlo simulations within the statistical error. Because of the absence of the negative sign problem, the VMC method can be applied to many different classes of quantum systems including weakly as well as strongly correlated electrons, frustrated quantum magnets, and interacting bosons.
Fig.: Low lying excitation spectra for a spin 1/2 anisotropic Heisenberg model on the triangular lattice.
Theoretical study on Ir oxides
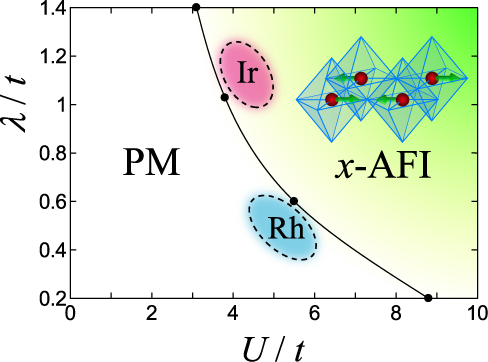 Sr2IrO4 is considered to be a novel Mott insulator induced by the large spin-orbit coupling, which is characteristic in 5d electron systems. To understand the mechanism of this spin-orbit-induced Mott insulator, we have studied the 3-orbital Hubbard model including the spin-orbit coupling term. Using the variational Monte Carlo method, we have obtained the ground state phase diagram (see Figure) and shown that the Coulomb interaction (U) and the spin-orbit coupling (λ) work cooperatively and induces the novel metal-Mott insulator transition. The total magnetic moment consisting of both orbital and spin components is ordered antiferromagnetically in the two-dimensional plane. Our result has also successfully described the metallic behavior of Sr2RhO4, a 4d counterpart with a larger U and a smaller λ.
Sr2IrO4 is considered to be a novel Mott insulator induced by the large spin-orbit coupling, which is characteristic in 5d electron systems. To understand the mechanism of this spin-orbit-induced Mott insulator, we have studied the 3-orbital Hubbard model including the spin-orbit coupling term. Using the variational Monte Carlo method, we have obtained the ground state phase diagram (see Figure) and shown that the Coulomb interaction (U) and the spin-orbit coupling (λ) work cooperatively and induces the novel metal-Mott insulator transition. The total magnetic moment consisting of both orbital and spin components is ordered antiferromagnetically in the two-dimensional plane. Our result has also successfully described the metallic behavior of Sr2RhO4, a 4d counterpart with a larger U and a smaller λ.
Fig.: The Ground state phase diagram of an effective 3-orbital Hubbard model with a spin-orbit coupling term. Transition from paramagnetic metal (PM) to spin-orbit-induced Mott insulator (x-AFI) is shown in U-λ plane.
Ultracold atomic/molecular gases in optical lattices
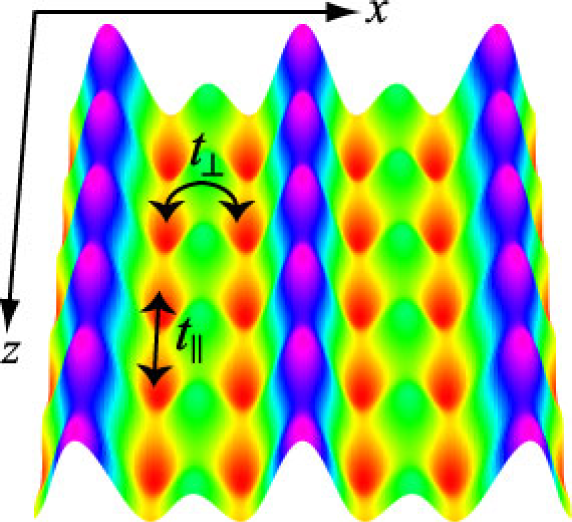 An optical lattice is created by interference of two counter-propagating laser beams, and it acts as a periodic potential for atoms. Systems of ultracold atomic/molecular gases are analogous to condensed matter systems such as electrons in solids and liquid helium in many aspects, and thorough studies of many-body physics in the optical-lattice systems will be useful for a deeper understanding of the condensed matter systems. Moreover, many properties in the optical-lattice systems can be widely and precisely controlled, including the lattice depth, the lattice structure, the interparticle interactions, and the spin degrees of freedom. This advantage allows us to explore novel phenomena (quantum phases, phase transitions, quantum transport, non-equilibrium dynamics, etc.) in parameter regions that have not been achieved in conventional condensed matter systems. We study the systems of cold gases confined in optical lattices by using several methods for quantum many-body systems, such as the time-evolving block decimation method, the variational Monte Carlo method, and Tomonaga-Luttinger liquid theory. Especially, we are interested in supersolids of dipolar gases (a gas that has a strong dipole-dipole interparticle interaction) and strongly correlated superfluids in low dimensional systems.
An optical lattice is created by interference of two counter-propagating laser beams, and it acts as a periodic potential for atoms. Systems of ultracold atomic/molecular gases are analogous to condensed matter systems such as electrons in solids and liquid helium in many aspects, and thorough studies of many-body physics in the optical-lattice systems will be useful for a deeper understanding of the condensed matter systems. Moreover, many properties in the optical-lattice systems can be widely and precisely controlled, including the lattice depth, the lattice structure, the interparticle interactions, and the spin degrees of freedom. This advantage allows us to explore novel phenomena (quantum phases, phase transitions, quantum transport, non-equilibrium dynamics, etc.) in parameter regions that have not been achieved in conventional condensed matter systems. We study the systems of cold gases confined in optical lattices by using several methods for quantum many-body systems, such as the time-evolving block decimation method, the variational Monte Carlo method, and Tomonaga-Luttinger liquid theory. Especially, we are interested in supersolids of dipolar gases (a gas that has a strong dipole-dipole interparticle interaction) and strongly correlated superfluids in low dimensional systems.
Fig.: Two-leg ladder lattice structure created with a double-well optical lattice.
Spin-dependent transport in nano-sized hetero-structures made of superconductors
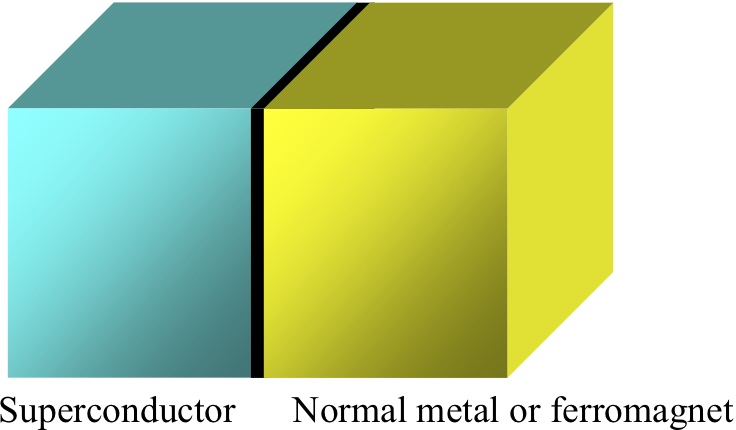
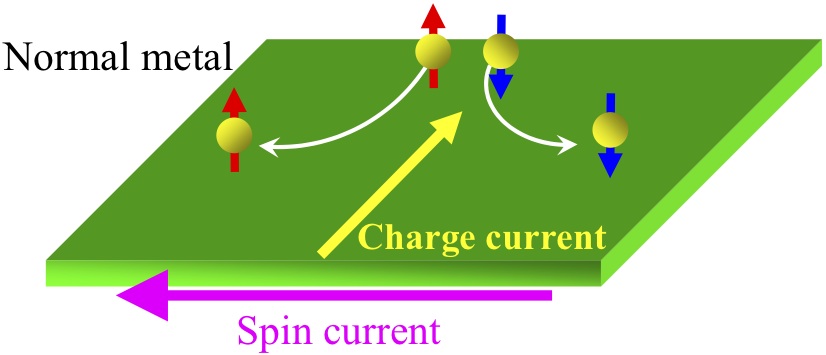 Fig. left: Schematic figure of a superconductor/normal metal or ferromagnet junction.
Fig. left: Schematic figure of a superconductor/normal metal or ferromagnet junction.
Fig. right: Schematic figure of the spin Hall effect. Charge current generates spin current and vise versa due to the spin-orbit coupling.
We develop a theory for spin-dependent transport in a nano-scale superconductor/normal metal or ferromagnet junction as shown in Fig. left. Specially, we focus on the spin Hall effect (Fig. right) and spin injection in these systems. Moreover, we propose new spintronics devices for which new physical phenomena occur in hybrid nano-structures based on superconductors. Theoretical methods we employ include non-equilibrium Green’s function and quasi-classical Green’s function techniques.
First-principles calculations of thermopower on metallic junctions
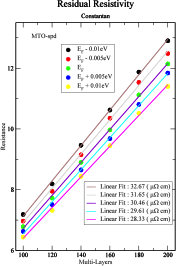 For a quantitative analysis on Peltier cooling effect on metallic junctions, the thermoelectric parameters for bulk as well as interfaces are estimated by first principles calculations based on the framework of density functional theory with a tight-binding muffin-tin orbitals basis. The Landauer-Büttiker formalism is used to study quantum transport coefficients, and the energy dependence of quantum scattering is determined based on the well-known Mott’s law. The resistivity of bulk CuNi (Constantan) alloy is given by the slope of the total resistance as a function of the thickness, which is estimated about 30.46 µΩcm at Fermi level (see Figure). The thermopower (=–S/T) at room temperature (T) is then estimated about 171.75 nV/K2. For CuNi/Au junction, we find that the estimated interface thermopower is around 100 nV/K2, which is significantly large compared to the bulk value. Therfore, we conclude that the interface contribution to the thermal transport in metallic junctions is much more important than one might have expected, and it needs to be considered for any quantitative comparisons with theoretical calculations and experimental observations.
For a quantitative analysis on Peltier cooling effect on metallic junctions, the thermoelectric parameters for bulk as well as interfaces are estimated by first principles calculations based on the framework of density functional theory with a tight-binding muffin-tin orbitals basis. The Landauer-Büttiker formalism is used to study quantum transport coefficients, and the energy dependence of quantum scattering is determined based on the well-known Mott’s law. The resistivity of bulk CuNi (Constantan) alloy is given by the slope of the total resistance as a function of the thickness, which is estimated about 30.46 µΩcm at Fermi level (see Figure). The thermopower (=–S/T) at room temperature (T) is then estimated about 171.75 nV/K2. For CuNi/Au junction, we find that the estimated interface thermopower is around 100 nV/K2, which is significantly large compared to the bulk value. Therfore, we conclude that the interface contribution to the thermal transport in metallic junctions is much more important than one might have expected, and it needs to be considered for any quantitative comparisons with theoretical calculations and experimental observations.
Fig.: Resistance for CuNi as a function of thickness of CuNi for different energy levels close to Fermi energy EF indicated in the figure.
Haldane-Anderson impurity model of transition metal-based proteins
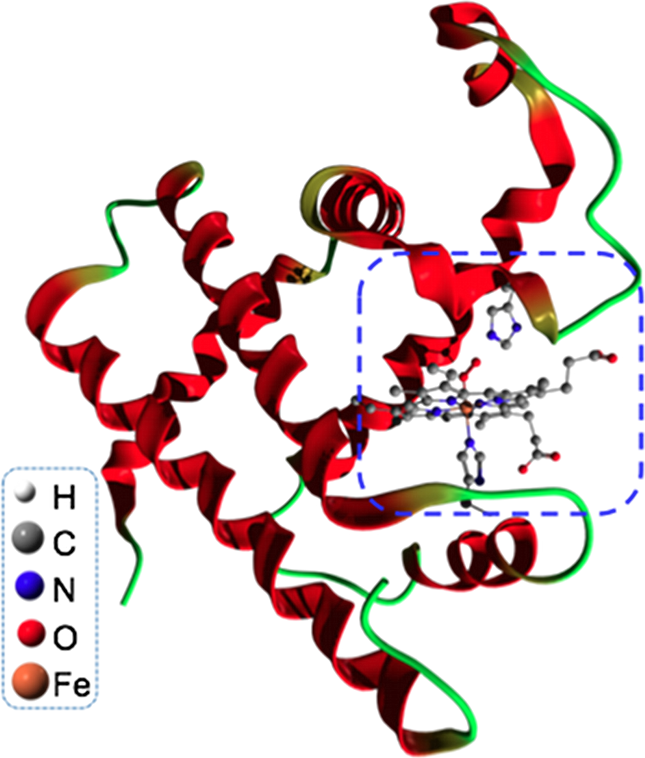 Heme-based metalloproteins are some of the most important proteins, central in biological processes as respiration or energy production (e.g. Myoglobin, Hemoglobin, Cytochrome P450). The biological activity of such protein is mainly determined by the spin state and the local charge of the transition metal localized at the center of the prophyrin active site. It is however well known that most common ab initio methods (e.g. DFT) usually predict an incorrect spin state for the ground state of many porphyrins, because the description of the electronic correlations of the transition metal are not adequate. In our project, we combine an ab initio DFT calculation and a quantum many-body model calculation. We use GAUSSIAN 03 to obtain, at the DFT level, the ab initio electronic ground state of a simplified myoglobin model, where a porphine group replaces the more complex active site, and an imidazol group replaces the proximal histidin. The effects of the bonding of biologically relevant molecules (e.g. O2 or CO) to the model myoglobin are investigated. Using these molecular "band", we build a Haldane-Anderson impurity model for the central transition metal ion which is coupled to the sorrounding molecules. To account correctly for the many-body effects in the prophyrin ring, we solve the 5-orbital Haldane-Anderson impurity model using various many-body techeniques such as quantum Monte Carlo methods and Density Matrix Renormalization Group methods.
Heme-based metalloproteins are some of the most important proteins, central in biological processes as respiration or energy production (e.g. Myoglobin, Hemoglobin, Cytochrome P450). The biological activity of such protein is mainly determined by the spin state and the local charge of the transition metal localized at the center of the prophyrin active site. It is however well known that most common ab initio methods (e.g. DFT) usually predict an incorrect spin state for the ground state of many porphyrins, because the description of the electronic correlations of the transition metal are not adequate. In our project, we combine an ab initio DFT calculation and a quantum many-body model calculation. We use GAUSSIAN 03 to obtain, at the DFT level, the ab initio electronic ground state of a simplified myoglobin model, where a porphine group replaces the more complex active site, and an imidazol group replaces the proximal histidin. The effects of the bonding of biologically relevant molecules (e.g. O2 or CO) to the model myoglobin are investigated. Using these molecular "band", we build a Haldane-Anderson impurity model for the central transition metal ion which is coupled to the sorrounding molecules. To account correctly for the many-body effects in the prophyrin ring, we solve the 5-orbital Haldane-Anderson impurity model using various many-body techeniques such as quantum Monte Carlo methods and Density Matrix Renormalization Group methods.
Fig.: Structure of myoglobin, protein responsible of muscle respiration. The active site (blue dash) is represented in ball-and-stick.
Variational cluster approximation based on the self-energy functional theory
 Variational cluster approximation (VCA) method based on the self-energy functional theory (SFT) takes into account precisely effects of short-range static and dynamical correlations, and thus superior to simple mean field approximations. The method has been applied successfully to a wide range of strongly correlated electronic systems to study, e.g., competition of various phases in high-Tc cuprates and in layered organic conductors, etc. Using this method, we have studied the ground state as well as low-lying excitations of 3d transition metal oxides (cuprates, Fe pnictides), 5d transition metal oxides (Sr2IrO4) and graphene to explore novel quantum states of strongly correlated electrons.
Variational cluster approximation (VCA) method based on the self-energy functional theory (SFT) takes into account precisely effects of short-range static and dynamical correlations, and thus superior to simple mean field approximations. The method has been applied successfully to a wide range of strongly correlated electronic systems to study, e.g., competition of various phases in high-Tc cuprates and in layered organic conductors, etc. Using this method, we have studied the ground state as well as low-lying excitations of 3d transition metal oxides (cuprates, Fe pnictides), 5d transition metal oxides (Sr2IrO4) and graphene to explore novel quantum states of strongly correlated electrons.
Fig.: One-particle excitation spectrum of Sr2IrO4.
New family of three-dimensional topological insulators with antiperovskite structure
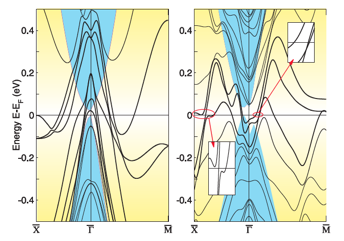 Up to now, all known topological insulators found experimentally and theoretically are related to two families with distinct crystal structures, i.e., one being a cubic non-centrosymmetric zinc-blende HgTe type and the other being a hexagonal centrosymmetric Bi2Se3 type. In this study, we have proposed a new family of materials for topological insulators in the antiperovskite structure. Through first-principles calculations, we have shown evidences that under a proper uniaxial strain, cubic ternary centrosymmetric antiperovskite compounds (M3N)Bi (M = Ca, Sr, and Ba) are three-dimensional (3D) topological insulators (figure). We have also discussed other related materials of the same antiperovskite structure, which are good candidates for 3D topological insulators. This proposed family of materials is chemically inert and the lattice structure is well matched to important semiconductors, which provides a rich platform to easily integrate with electronic devices
Up to now, all known topological insulators found experimentally and theoretically are related to two families with distinct crystal structures, i.e., one being a cubic non-centrosymmetric zinc-blende HgTe type and the other being a hexagonal centrosymmetric Bi2Se3 type. In this study, we have proposed a new family of materials for topological insulators in the antiperovskite structure. Through first-principles calculations, we have shown evidences that under a proper uniaxial strain, cubic ternary centrosymmetric antiperovskite compounds (M3N)Bi (M = Ca, Sr, and Ba) are three-dimensional (3D) topological insulators (figure). We have also discussed other related materials of the same antiperovskite structure, which are good candidates for 3D topological insulators. This proposed family of materials is chemically inert and the lattice structure is well matched to important semiconductors, which provides a rich platform to easily integrate with electronic devices
Fig.: Electronic band structures for (001) surface of (Ca3N)Bi with spin-orbit interaction. Left: no strain applied. Right: 7% stretching strain applied in ab-plane while keeping the c-lattice constant fixed.
Kernel Polynomial Method on GPU
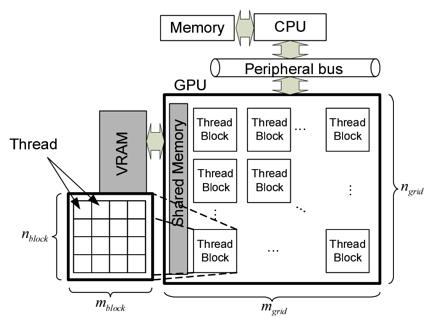 Kernel Polynomial Method (KPM) is an effective numerical method for evaluating eigenvalues of a huge Hamiltonian matrix. Applying Chebyshev polynomials and appropriate kernel functions to KPM, one-particle as welll as two-particle dynamical correlation functions are effectively estimated with controllable accuracy. Regarding the performance of KPM, massive parallelism on Graphics Processing Units (GPU) or supercomputers for accelerating the algorithm has a special meaning for treating much larger systems. In our numerical experiment, the KPM implementation on GPU shows more than 10 times faster compared with CPU. The target of this research is to build easy-to-use libraries which can be used in GPU or CPU cluster environments for helping solve grand challenges.
Kernel Polynomial Method (KPM) is an effective numerical method for evaluating eigenvalues of a huge Hamiltonian matrix. Applying Chebyshev polynomials and appropriate kernel functions to KPM, one-particle as welll as two-particle dynamical correlation functions are effectively estimated with controllable accuracy. Regarding the performance of KPM, massive parallelism on Graphics Processing Units (GPU) or supercomputers for accelerating the algorithm has a special meaning for treating much larger systems. In our numerical experiment, the KPM implementation on GPU shows more than 10 times faster compared with CPU. The target of this research is to build easy-to-use libraries which can be used in GPU or CPU cluster environments for helping solve grand challenges.
Fig.: Execution Model of CUDA