Ab initio phasing
RosettaX
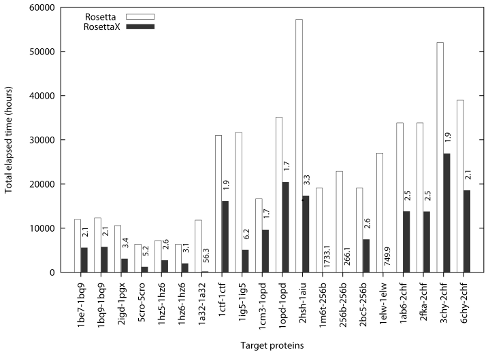
The ability to predict protein structures from sequences has opened up many new possibilities. Although the accuracy of predicted models are not sufficient for structure-based drug design, however, one utility of the predicted models is to be used as templates to solve the protein crystallographic phase problem by molecular replacement. This is the so called “ab initio phasing by de novo models” method. Typically, the procedure in this method is to first generate several hundred thousand models and select a few hundred lowest energy models for molecular replacement. We have modified this procedure by embedding the molecular replacement during the generation of each model. Once a few good models have succeeded in molecular replacement, the entire simulation is terminated, thereby saving a large amount of computing time. Our method is termed RosettaX
(publication).
RosettaX is shown to be ~140 times faster for these targets on average.